Ethics and Institutional Review Board applications
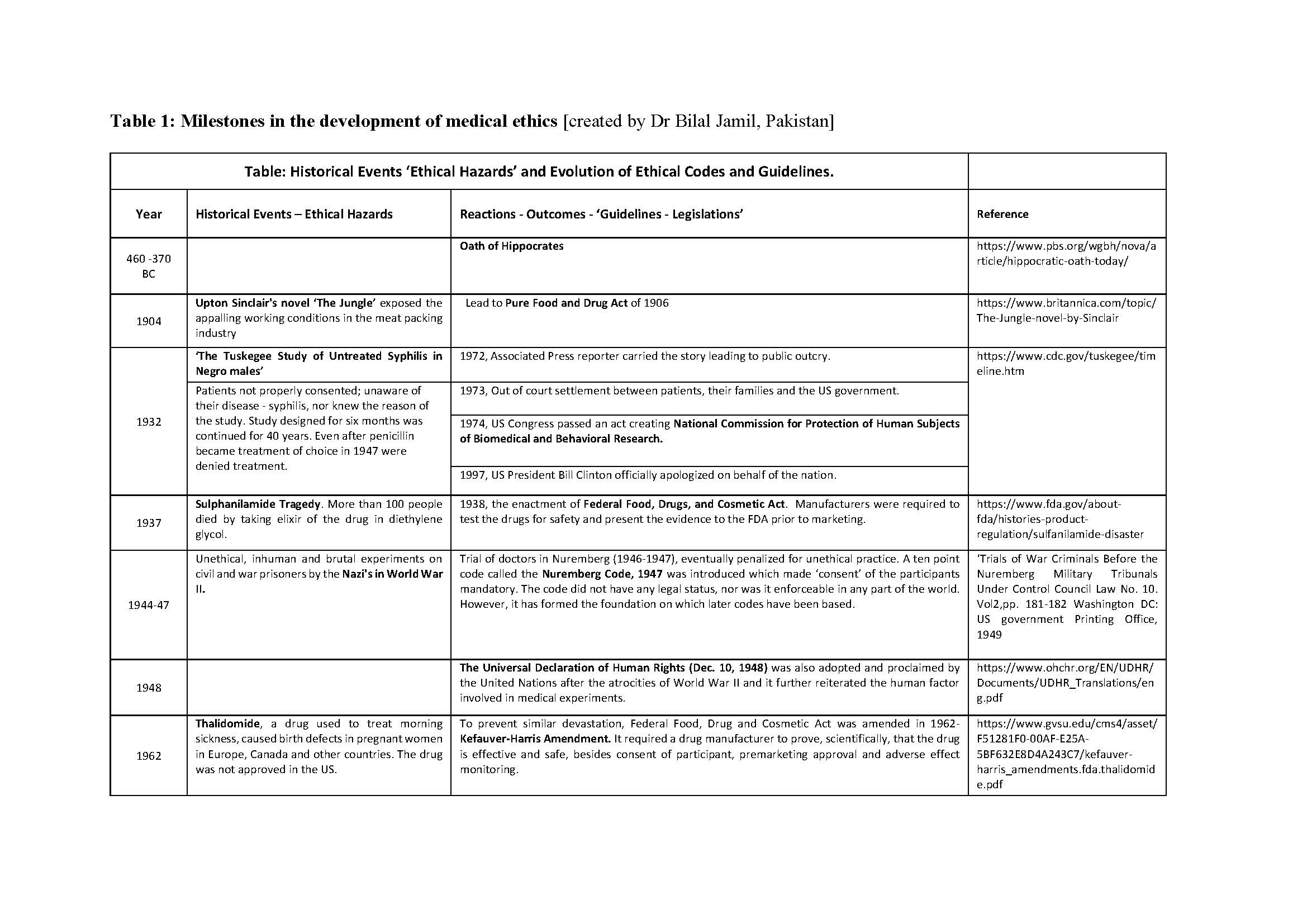
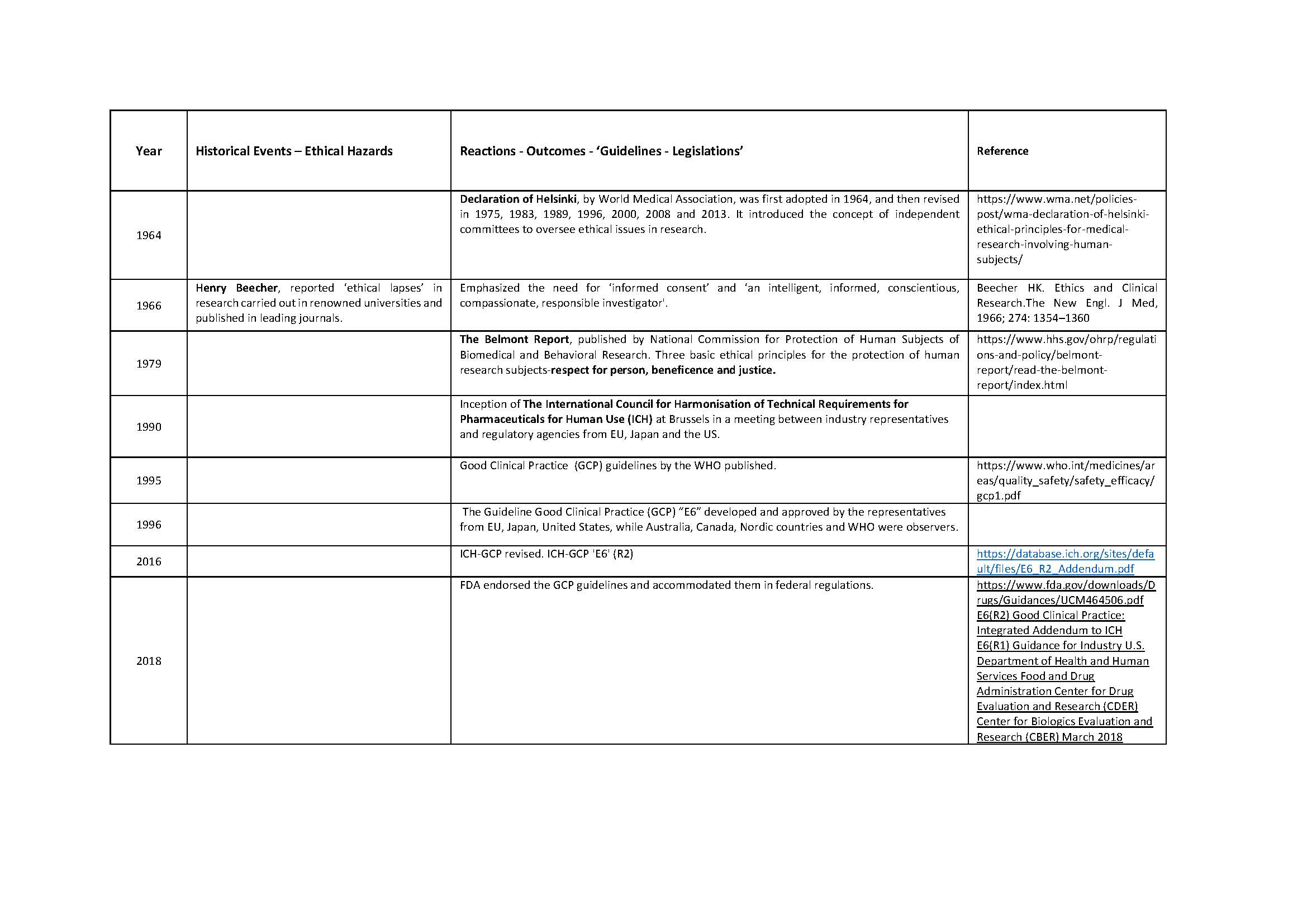
Medical ethics is a set of values that applies to the practice of clinical medicine and in scientific research. Hippocrates (460-370 BC), the father of medicine was the first to define them in the Hippocratic Oath. Similarly, the dictum ‘Primum non nocere’ (Latin) ‘First do no Harm’ is centuries old. Despite this, history is testimony to the fact that these ‘oaths’ have often been ignored in human research. Edward Jenner (1749-1823) tested small pox vaccine on his son, and neighbors. James Marion Sims (1813-1883) researched on enslaved black women. The Tuskegee Study of Syphilis in Negro Males (1932-1972) was conducted on subjects who were kept in dark about their disease, about the purpose of the study, and were deprived of treatment once it was available. Inhuman and brutal experiments were done, by the Nazis, on the prisoners during the Second World War (1939-1945). It was not just the under privileged or marginalized who suffered. Henry Beecher reported ‘ethical lapses’ in research carried out in some renowned Universities in the developed world. The research was published in leading journals. Similar events of the past and their reverberations are shown in Table 1. Tragically, healthy volunteers have also suffered and even died during clinical research; events which, unfortunately, are not only of historical interest [Nature, 2016].
Principles of Medical Ethics
Medical ethics is based on four prima facie principles. Respect for autonomy, beneficence (doing good), non-maleficence (not doing any evil or harm), and justice. They offer a basic common analytical framework and a common basic moral language. Respect for autonomy acknowledges the dignity and freedom of every person. It requires obtaining informed consent from research subjects, or their legally authorized representatives. Beneficence and non-maleficence requires researchers to maximize benefits and minimize harms during research. Research-related risks must be reasonable in light of the expected benefits, or simply must provide net benefit to the patient. In the setting of clinical trials, it implies as no patient should suffer as a result of participating in a trial and no patient should be denied known effective treatment. However, known effective treatment relies on objective evidence and not on subjective belief. Justice requires equitable selection and recruitment and fair treatment of research subjects.
The Role of Institutional Review Boards/Ethics Committees
To ensure medical research is ethical, it is prudent that institutions involved in research, especially involving human subjects, are overseen by a formally designated, independent, group of trained professionals. These professional committees are commonly called Institutional Review Board (IRB) or Ethical Review Board (ERB). Their role is to ensure that all research performed at that institution upholds the principles of medical ethics such that participants are protected from undue risks.
To seek an IRB’s approval, one has to submit a written proposal of the planned research. Before doing so, go through the guidelines of the relevant IRB. A research proposal should generally include:
Title
A short title which describes the key proposed questions.
Preamble
This should discuss the background and include a review of literature highlighting key debates and developments to rationalize the need for the proposed study. Any previous, similar or related research involving either animals or humans should also be summarized.
Research Question
The objectives of the research should be described. What questions and issues would be explored? The need and appropriateness of the study should be justified.
Selection of participants
Participants’ entry criteria should be detailed. Criteria for age, gender, or any other factors should be included. Method of recruiting participants for research should be described. Exclusion criteria should also be mentioned.
Consent
Informed consent should describe, in simple non medical terms what the research is about, what is expected from the participant, what benefits are expected from the study, and what is the risk involved. It should also clarify that the participation is voluntary and that a participant may withdraw from the study any time at his free will without any fear or penalty. How the confidentiality and anonymity would be protected must also be explained.
Research Methodology
Study design must be clearly outlined. Randomization and control methods should be elaborated. The investigational activities, treatments, or procedures must be detailed. Endpoints must be defined. Study data collection, analysis and its interpretation should be detailed including analysis of data during the study. Advantages and limitations of the research approach should be discussed.
Plan of work and time frame
Outline of various stages of the study with a timeline should be mentioned.
Monitoring Adverse Effects
Methods of monitoring, documenting and managing adverse events should be mentioned. In case of adverse effects or for any other reason criteria for stopping the trial should be stated.
Data Management and Interpretation
The method for analysis of the data should be described.
Resources
Availability of resources required for the research including qualified personnel, their training, equipments or any other must be documented and confirmed.
References
All references cited should be properly listed.
Note that many organizations have standardized application forms, typically submitted online.
Depending on the risk the participants would be exposed to. A research proposal may fall into one of three categories: Exempt of review, Expedited review, or a Full Board review.
Exempt of review:
‘Exempt’ means review by one IRB member, sometimes in consultation with another. A research activity may be declared exempt if it is considered low-risk e.g.
- Research conducted in established or commonly accepted educational settings, involving normal educational practices.
- Research using anonymous or no-risk tests, surveys, interviews, or observations.
- Most research involving public officials.
- Research involving the collection or study of existing data if it is publically available or if subjects cannot be identified.
- Research examining public benefit or service programs.
- Taste and food quality evaluation and consumer acceptance studies
Expedited review:
Research may qualify for ‘expedited review’ if it involves only minimal risk to participants, does not include intentional deception, does not employ sensitive populations or topics, and includes appropriate informed consent procedures. For example, the collection of physical data through non-invasive procedures is eligible for an expedited review, including: Height and weight, ECG, MRI, Ultrasound, moderate exercise, blood or other body fluids. ‘Expedited Review’ is usually done by the IRB chair and one or more experienced reviewers.
Full board review:
A full board review is required for research that is judged to involve more than minimal risk, or involves protected populations such as children, prisoners, or disabled individuals. The following categories of research require full IRB approval:
- Projects for which the level of risk is determined by the IRB Chair to be greater than minimal.
- Projects that involve the intentional deception of subjects, such that misleading or untruthful information will be provided to participants.
- Projects that involve sensitive or protected populations (such as children or cognitively disabled individuals).
- Projects that plan to use procedures that are personally intrusive, stressful, or potentially traumatic (stress can be physical, psychological, social, financial, or legal).
Time taken by the IRBs may vary depending upon the category of review and schedule of the IRB. Investigator should check this with the relevant IRB in advance.
The number and structure of IRBs varies widely between different jurisdictions. In some countries, agreements have been reached such that at IRB approval at one site can be held to cover multiple other sites in the same country. In other countries, a central screening application to a national body is required prior to submitting to the local IRB representing the study site.
- ClinRegs [very useful site aggregating links to IRB and regulatory information around the world, maintained by US NIH]
- Council for International Organizations of Medical Sciences (CIOMS): International ethical guidelines for health-related research involving humans
- Research in Low-Resource Countries: Ethical Review [Nuffield Council on Bioethics]
- Declaration of Helsinki – Ethical Principles for Medical Research Involving Human Subjects
- Office for Human Research Protections [portal linking to guidance on US regulatory requirements, including ethical review; US Dept. of Health & Human Services]
- Gillon, PR. Medical Ethics: Four principles plus attention to scope. BMJ. 1994;309:184–188. [overview of principles of medical ethics]
- ICH Good Clinical Practice guidelines [includes descriptions of the roles of investigators and sponsors, including as these related to ethics and consent]
- Institutional Review Board (IRB) Written Procedures: Guidance for Institutions and IRBs [want to know what US IRBs are required to do? Here is the summary from US Dept. of Health & Human Services]
- IEA online textbooks [includes Field Trials of Health Interventions\Ethical considerations]