
December 2023 Edition

Genetics has emerged as a transformative force in nephrology, unlocking the complex genetic codes that shape kidney health. This curated selection of research papers from ISN Journals presents the current landscape of gene exploration in kidney care, offering new insights into the role of genetic markers, pathways, and personalized interventions.
KIDNEY INTERNATIONAL ARTICLES |
A Wave of Deep Intronic Mutations in X-linked Alport Syndrome
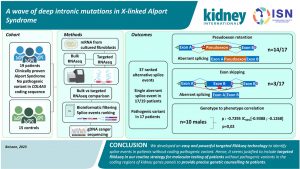
Should targeted RNA sequencing be a part of routine molecular testing for X-linked Alport syndrome?
Alport syndrome is an X-linked disorder with a defect in the COL4A3-5 coding sequence. This causes glomerular basement membrane disruption. Identifying splice events in those who lack specific coding for the pathogenic variant of this disease process may be helpful. With proper detection of such monogenic disease, targeted therapy on deep-intronic variants may eventually allow for adequate treatment/management. However, events like nonsense-mediated decay as a result of splicing may cause some limitations. Should targeted RNA sequencing be part of routine molecular testing where patients without pathogenic variants in the coding regions can be identified? This may contribute to effective genetic counselling for patients. It will also be important in kidney donor selection.
The Axolotl Kidney: A Novel Model To Study Kidney Regeneration

Chronic kidney disease remains a significant public health burden worldwide, with no optimal treatment options other than dialysis or kidney transplantation. Kidney regeneration is now a possible treatment option for people with kidney failure. In the case of minor injury, the mammalian kidney can partially repair damaged nephrons and, in the case of severe injury, can progress to irreversible renal fibrosis.
On the other hand, the axolotl (Ambystoma mexicanum) has the incredible ability to regrow various bodily components, including entire limbs, tail, ocular tissues, liver, heart, and even brain. The axolotl kidney shares morphological, molecular, and functional similarities with the mammalian kidney. This study is the first to present information on axolotl kidney regeneration after injury and regeneration in two different nephrotoxicity models.
In the gentamicin-induced nephrotoxicity axolotl nephrons model, the renal tubules regenerate two months after severe renal injury. The bulk RNA sequencing data revealed overexpressed genes associated with cell proliferation, extracellular matrix remodeling, and nephrogenesis. The activation of nephrogenesis-related genes such as Sox9 and Wnt4 during axolotl kidney regeneration suggests that their function has been conserved over time.
The second model, doxorubicin-induced glomerular injury, demonstrated that four weeks after injury, glomeruli were regenerated and much smaller than those in the control group. The size of the regenerated glomerulus is comparable to that of the control group 8 weeks following injury. Podocin expression was also restored while the glomerular regeneration continued.
A Novel Multi-ancestry Proteome-wide Mendelian Randomization Study Implicates Extracellular Proteins, Tubular Cells, And Fibroblasts iIn Estimated Glomerular Filtration Rate Regulation.
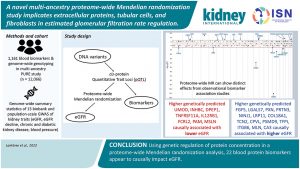
Is there any relationship between biomarkers and the estimated glomerular filtration rate regulation? In this prospective observational study, Lankre et al., searched for genetic changes predictive of the concentration of more than 1000 biomarkers and found that 22 different blood biomarkers changed kidney function. Could these findings be the watershed for novel therapeutic targets?
On the Relevance of Thrombomodulin Variants In Atypical Hemolytic Uremic Syndrome

Atypical hemolytic uremic syndrome (aHUS) is a potentially lethal medical condition often resulting in chronic kidney disease. Genetic testing is critical in clinical care because pathogenic variants in complement genes account for 50% to 60% of cases of aHUS. Their identification reinforces diagnosis and assists clinical decisions, such as treatment with anti-complement drugs. One gene of interest is THBD, which encodes thrombomodulin (TM), an anticoagulant endothelial membrane glycoprotein that regulates complement by binding to factor H and accelerating the inactivation of C3b.
The aHUSC3G registry is a repository of clinical and research data on Spanish aHUS and C3G patients established to facilitate analysis by researchers. Huerta et al. report on the genetic and clinical retrospective study of 27 patients from this registry carrying THBD variants.
The authors found that in this cohort, none of the patients carrying isolated THBD variants had a disease recurrence throughout the entire follow-up, including four patients who received a kidney graft without preemptive eculizumab. Most patients carrying isolated THBD variants had ≥1 cause associated with secondary HUS and tended to recover spontaneously after removing the putative underlying cause.
Overall, the conclusion was that patients carrying THBD variants do not exhibit the prototypical clinical profiles of patients with complement-mediated aHUS and resemble those with secondary HUS. This information is crucial for their therapeutic management and raises the issue that THBD variants found in aHUS patients may merely reflect their prevalence in the normal population.
Rare Variants in the Sodium-Dependent Phosphate Transporter Gene SLC34A3 Explain Missing Heritability of Urinary Stone Disease
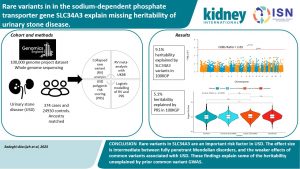
Although the etiology of urinary stone disease (USD) is multifactorial, genetics plays a significant role in its pathogenesis; it is estimated that the heritability of USD is around 45%. In this investigation, Sadeghi-Alavijeh et al. look for the presence of the SLC34A gene, which encodes the sodium-dependent phosphate transport protein 2C expressed in the proximal tubule (NaPi-IIc) in people with kidney stones, and found changes in 1 of 2 copies of the SLC34A3 gene (present in 5% cases compared with 1.6% of controls).
The mechanism by which changes in SLC34A increase the risk of USD is due to a reduction in proximal tubular phosphate transport.
Repurposing Small Molecules for Nephronophthisis and Related Renal Ciliopathies
Nephronophthisis and related renal ciliopathies (NPH-RC) are a group of autosomal recessively inherited disorders arising from variants in the gene coding for primary cilium components.
Over the past two decades, advances in understanding disease mechanisms have identified several dysregulated signaling pathways, some shared with other cystic kidney diseases.
In this review, the authors demonstrate how a better understanding of disease mechanisms allowed a knowledge-based approach for repurposing molecules previously developed against specific pathways in mouse models. They also discuss how unbiased “in cellulo” phenotypic screens identified small molecules which could be used to rescue ciliogenesis defects seen in NPH-RC.
KIDNEY INTERNATIONAL REPORTS ARTICLE |
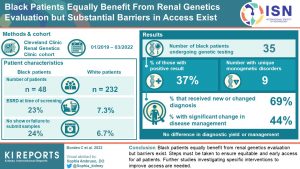
Barriers to accessing genetic evaluation has been shown to be higher in Black patients.
More than 600 genes have been closely associated with kidney disease, ranging from polycystic disease to hereditary nephritis that specifically affects genes.
Knowledge of genetic analysis is an advancement in nephrology, which helps with proper diagnosis and appropriate management.
A study by researchers in Canada demonstrates racial parity in the genetic evaluation of these patients. The authors discuss the barriers and measures that can be taken to improve access to genetic testing.
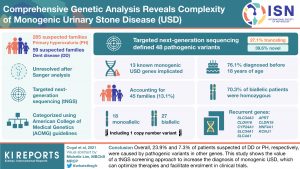
Genetic screening in patients with urinary stone diseases (USD) or nephrocalcinosis (NC) is valuable in characterizing the defective gene, guiding management, informing family screening and conducting clinical trials.
Two well-characterized monogenic causes of USD and NC are primary hyperoxaluria (PH) and Dent disease (DD). In this study, the authors use targeted next-generation sequencing (tNGS) to rescreen a cohort of patients clinically suspected of having PH or DD but unresolved from targeted Sanger analysis for known PH and DD genes (AGXT, GRHPR, HOGA1, CLCN5, OCRL).
The cohort included 285 unresolved patients clinically suspected of PH(PHN) and 59 suspected of DD (DDN). They used tNGS panels of 90 genes (n=279) and 102 genes (n=65) containing known monogenic USD/NC genes, as well as candidate genes important for calcium metabolism or urinary components that contribute to lithogenicity, including oxalate, citrate, uric acid, or pH.
From this analysis, a likely cause of the disease due to variants in known monogenic USD genes was identified in 45 families (13.1%), 29 PHN (10.2%), and 16 DDN (27.1%). Of the resolved cases, 27 (60%) had biallelic disease and 18 (40%) had monoallelic disease, with 14 different genes implicated overall. Of the 48 defined pathogenic variants, 19 (39.6%) were novel and 13 (27.1%) truncated. The most common mutated genes were CLDN16 and SLC34A3.
The study highlights the phenotypic overlap across monogenic USDs and shows the value of a tNGS screening approach to improving diagnosis. This approach could help to optimize therapies and facilitate enrollment in clinical trials.

Thrombotic microangiopathy (TMA) presents a multifaceted range of syndromes, each reflecting profound endothelial injury triggered by diverse mechanisms. Among these, the pivotal involvement of the complement system surfaces prominently in a subset of TMA patients. Recognizing this connection assumes paramount clinical significance as it not only elucidates the intricate pathogenesis but also directs the course and duration of treatment strategies. Understanding the interplay between TMA and the complement system unveils critical insights that serve as a compass, guiding clinicians toward more targeted and productive therapeutic interventions tailored to the distinct needs of affected individuals.
This work by Sjoerd and colleagues looks at a well-defined cohort of patients with TMA and hypothesizes that assessing serum-induced ex vivo C5b9 formation on the endothelium and screening for rare variants in complement genes can better categorize TMA.
Ex vivo C5b9 formation was found in all patients with primary atypical hemolytic uremic syndrome and in 59% of patients with TMA and coexisting conditions. Ex vivo C5b9 formation was associated with rare genetic variants vs. patients with normal ex vivo C5b9 formation. They concluded that ex vivo C5b9 formation and genetic testing appear to categorize TMAs into different groups because they identify complement as a driving factor of disease, with potential therapeutic and prognostic implications.
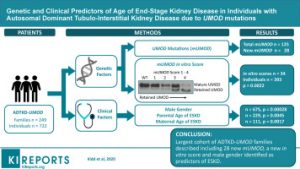
Autosomal dominant tubulointerstitial kidney disease caused by UMOD mutations (ADTKD UMOD) is a rare syndrome with considerable variability in the age of end-stage kidney disease (ESKD). The minor allele of rs4293393, situated in the promoter of the UMOD gene, is prevalent in 19% of the population and reduces uromodulin production by approximately 50%, perhaps affecting the age of ESKD.
The genetic and clinical variables linked with the age of ESKD onset were investigated in this study. An in vitro score of mUMOD transit and the presence of gout, age of gout onset, and parental age of ESKD were all predictors of ESKD onset. The rs4293393 UMOD minor allele, which is linked to lower uromodulin production, was shown to be under-represented in ADTKD-UMOD families.
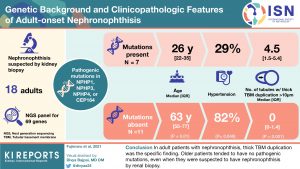
Nephronophthisis (NPH) has recently been considered a monogenic cause of end-stage kidney disease (ESKD) in adults. In this study, the authors looked at 18 sporadic adult patients suspected of having NPH by renal biopsy. They analyzed 69 genes that cause hereditary cystic kidney disease and compared clinicopathologic findings between patients with and without pathogenic mutations in NPH-causing genes.
Of the 18, only 7 had pathogenic NPH-causing mutations. These patients were younger than those without mutations (median age, 26 vs. 63 years), and none were > 50 years of age. They were more likely to be male (71% vs 9%, p= 0.01) with less hypertension (29 vs 82%, p= 0.049). While classic NPH findings were similar, the number of tubules with thick basement membrane duplication was higher in those with a pathogenetic mutation. Thus, the results of this small study suggest some clinicopathological features that may help classify adults with NPH features on biopsy.